近日,我院杨勇教授和程俊教授研究团队以P2-Na2/3(Mg1/3Mn2/3)O2为研究对象,首次联用密度泛函理论 ( DFT) - 基于机器学习的深度势能分子动力学模拟(DPMD)计算23Na NMR化学位移,确认了该体系中实验观测到的两个23Na化学位移峰分别来源于两种层间堆垛方式(空间群 P63/mcm和 P6322 ),修正了人们之前一直认为该类材料仅有单一堆垛(P63/mcm) 的认知。相关研究结果“Unravelling the Fast Alkali‐ion Dynamics in Paramagnetic Battery Materials Combined with NMR and Deep‐Potential Molecular Dynamics Simulation”发表在Angew. Chem. Int. Ed.(2021, doi: 10.1002/anie.202102740)。
P2 型锰基钠离子正极材料因元素的丰富性及其安全无毒,被认为是理想的大规模储能材料。该类层状氧化物材料具有较大的层间距可允许 Na+ 的快速嵌入/脱出,所呈现的优异倍率性能吸引了广泛的研究。核磁共振谱作为一种无损且对局域结构敏感的分析表征手段,可获取多种电池材料的(局域)结构与离子扩散动力学等信息。然而,因 P2 型材料结构中 Na+ 的移动速度很快,且存在过渡金属离子与被观测核复杂的相互作用,使得人们对其获得的核磁实验谱峰难以准确指认。
课题组采用基于深度神经网络的机器学习势函数并运用其进行分子动力学模拟,即深度势能分子动力学 (DPMD)。 DPMD不仅可以达到与常规 DFT 计算相同的计算精度,还可以加快约百万倍的计算速度。对于P2-Na2/3(Mg1/3Mn2/3)O2,常温下 200 纳秒的 DPMD 模拟才能得到 Na+ 收敛的精确占位,据其计算得到的 NMR 位点平均位移很好的符合了实验测量值,得以指认低位移峰对应P63/mcm,高位移峰对应 P6322,且确定了P6322 的含量为 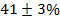 。DPMD模拟预测了Na+在中P6322中的扩散更快,这点也被 23Na 核弛豫时间的测量所确认。
。DPMD模拟预测了Na+在中P6322中的扩散更快,这点也被 23Na 核弛豫时间的测量所确认。
通过构效关系分析, 课题组认为P6322的电化学循环性能略低于P63/mcm 型材料,因为深度脱钠后层间的“-O-O-”对所处环境的对称性较低,可能会导致其偏向相邻层间的Mg或者Mn,这将不利于后续氧反应的可逆性。该工作还可启发我们可以通过减少 P6322 的含量来调控P2-Na2/3(Mg1/3Mn2/3)O2 电化学性能,结合DPMD计算化学位移的方法,适用于目前层状金属氧化物材料中所存在的快离子运动体系。

论文共同第一作者是我院博士研究生林敏和刘湘思,该工作得到了美国国家强磁场实验室傅日强教授的帮助。研究工作得到国家自然科学基金重点项目 (219350092),电化学研究方法创新群体项目(22021001) ,以及国家重点研发计划课题( 2016YFB0901502、2018YFB0905400) 的支持。
论文链接: https://doi.org/10.1002/anie.202102740

